





Energy calculations are faster in protein design
Computer-aided development of new proteins for biomedical or other applications requires long computing times on high-performance servers. A team of researchers from the Max Planck Institute for Biology in Tübingen and the University Hospital in Tübingen has developed and tested a new computational method that dramatically speeds up the necessary energy calculations. Their approach enables the accurate and efficient development of functional proteins. The researchers also demonstrated the usefulness of their findings by developing two classes of proteins that could be used in the diagnosis and treatment of cancer.
© Katerina Maksimenko
Protein design involves several interaction energy calculations.
© Katerina Maksimenko
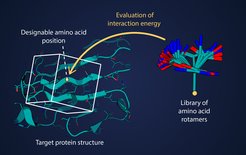
Proteins are vital in many respects: some ensure strong hair and bones, others regulate metabolism or recognize and fight viruses – just to name a few of their various tasks. Therefore, the development of new proteins with specific functions has the potential to revolutionize biomedicine.
When researchers design a protein with a desired function, they first determine its overall spatial structure, because shape determines how the protein interacts with other molecules. The challenge then is to find a consistent arrangement of building blocks from which the target structure can be assembled. These building blocks are amino acids: organic molecules linked together in long chains to form proteins. The interaction of amino acids and all of their atoms determines the three-dimensional shape of a protein and thus its function.
Radically reduced computational effort
And since the sequence of amino acids in a protein determines the shape, even small changes — such as replacing one amino acid with another — can dramatically stabilize or destabilize the desired structure. “In order to develop a stable protein, you have to calculate and compare the energies of countless amino acid sequences,” explains Mohamed Elgamsy, head of the research group at the University Hospital in Tübingen. “From a computational point of view, these calculations are a bottleneck for protein design.”
Therefore, El-Gamasy and his collaborators have now developed a new computational method that not only speeds up energy calculation exponentially, but also allows for fundamental improvements in accuracy. They were the first to apply the technique known as stretching to a protein design problem, a method already widely used in rendering graphics and computer games.
Instead of calculating the interaction energy for each pair of spatially close atoms individually, they pre-select most of the reaction information for each amino acid and store it in a 3D grid. Using these networks, the reaction energies can easily be calculated for each case. “We are particularly pleased that energy calculations can be accelerated without machine learning,” says Katerina Maksymenko, a researcher in the Department of Protein Evolution at the Max Planck Institute for Biology in Tübingen and lead author of the study. “Our calculations are purely based on physics. Therefore, we can always understand how and why the program achieved its results. That would be impossible with a deep learning algorithm.
Clinical applications in the diagnosis and treatment of cancer
The researchers also demonstrated the benefits of their method: they developed and tested two classes of proteins in collaboration with researchers from the University Hospital Tübingen and the Werner Siemens Imaging Center. The first invention blocks communication between receptors for EGF, a protein that regulates cell growth and differentiation and plays an important role in various types of cancer.
The newly developed second protein is also suitable for direct clinical use. “What’s unique about this is the potential dual effect on cancer diagnosis and treatment,” Maksimenko stresses. “The protein binds to copper-64; that is, its distribution in the body can be traced using nuclear medicine. But it can also transport the radioisotope copper-67, which kills cancer cells. In addition, the protein can bind to 13 copper ions at the same time; this density is inconsistent. Its precedents make the molecule particularly suitable for imaging at the highest quality.
In a further development step, the team wants to associate the copper bond with other relevant therapeutic proteins and investigate their distribution in the body. The researchers are convinced that their approach will support the development of many other proteins.

“Alcohol buff. Troublemaker. Introvert. Student. Social media lover. Web ninja. Bacon fan. Reader.”
More Stories
Weather: Strongest solar storm since 2003 triggers Swiss northern lights
Where is this natural lake located that has been hidden for thousands of years?
Solar storms and northern lights: what they mean for Earth